
논문 리뷰 — AlphaFold 3: Accurate Structure Prediction of Biomolecular Interactions
Published:
원문: Abramson et al., “Accurate structure prediction of biomolecular interactions with AlphaFold 3”, Nature 630, 493–500 (2024) DeepMind + Isomorphic Labs. 2024년 5월. 코드: google-deepmind/alphafold3 (2024-11 비상업 공개, 2025-02 일반 공개, CC-BY-NC-SA)
2024 노벨 화학상(Hassabis, Jumper)의 근거가 된 논문. AF2가 단일 단백질 접힘을 실험 오차 수준까지 풀어버린 게 2020–21년이었고, AF3는 그 이야기를 리간드·DNA·RNA·이온·공유변형을 포함한 복합체로 확장했다. “All of life’s molecules”라는 과장스러운 카피가 가리키는 게 바로 이 지점.
AF2 논문을 아는 사람 기준으로, 뭐가 남고 뭐가 바뀌었는가 정도만 본다.
왜 AF2로는 부족했나
“단백질은 풀렸다”는 수식어가 AF2에 붙긴 했지만, 생물학에서 의미 있는 사건은 거의 다 복합체에서 일어난다. 항체가 항원에 붙고, 전사인자가 DNA를 만나고, 약이 포켓에 들어간다. AF-Multimer가 단백질-단백질 복합체까지는 연장했지만 저분자·핵산·이온은 여전히 밖이었다. 약을 만드는 쪽에서 보면 반쪽짜리 도구였던 거다.
AF3는 이걸 한 모델에서 한 번의 추론으로 같이 푼다. 그게 된다, 그것도 꽤 잘.
구조 — 세 개가 바뀌고, 하나가 남았다
AF2: MSA + Templates → Evoformer → Structure Module (회귀) → 좌표
AF3: 원자 그래프 → Pairformer → Diffusion Module → 좌표
(recycling은 양쪽 다 있음)
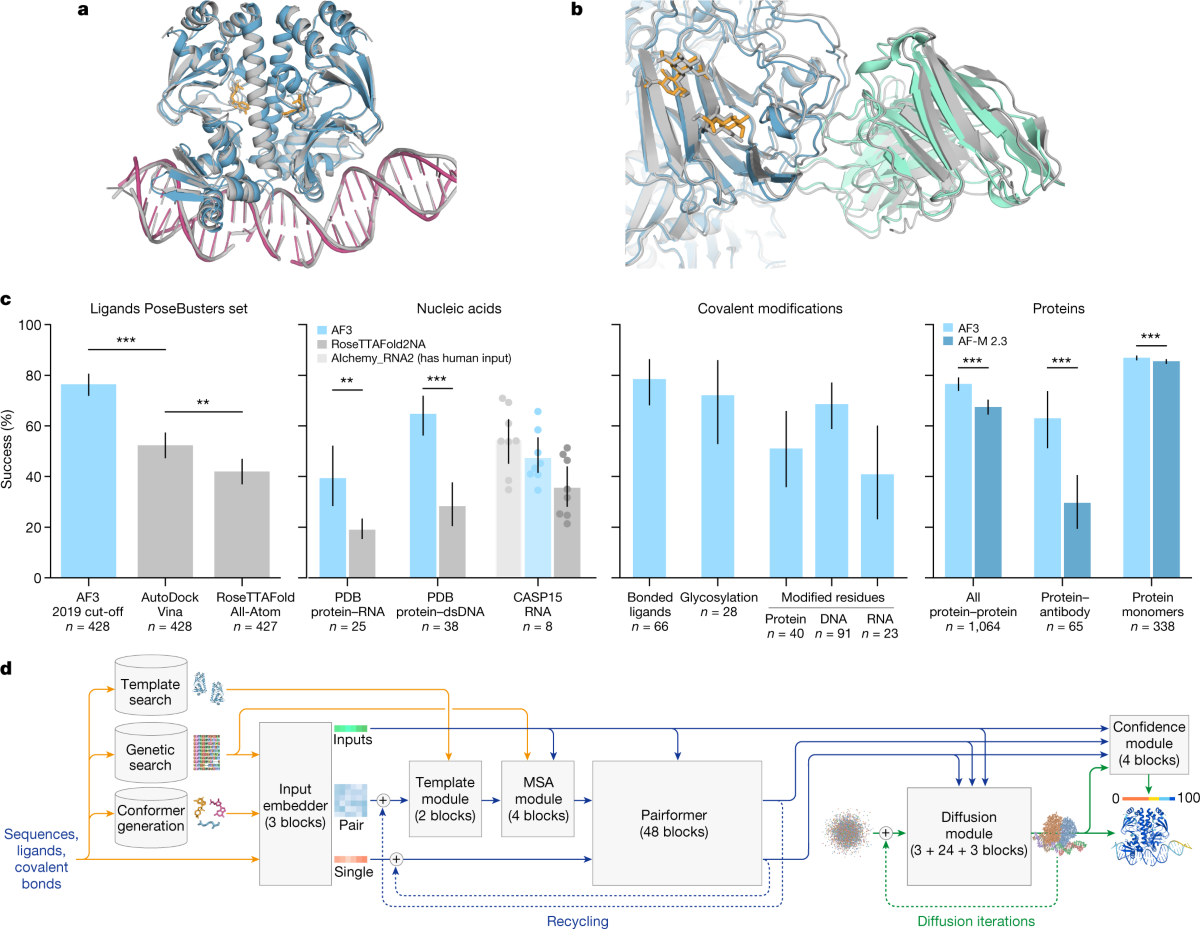 Fig. 1 (Abramson et al., 2024). 상단 — AF3가 예측한 단백질-DNA, 항원-항체 복합체 예시. (c) 리간드·핵산·공유 변형·단백질 각 카테고리에서 AF-M 2.3, RFAA, AutoDock Vina 등 베이스라인과의 비교. (d) 전체 파이프라인: 입력이 Pairformer로 들어가고, 그 pair representation을 조건으로 Diffusion Module이 원자 좌표를 생성한다.
Fig. 1 (Abramson et al., 2024). 상단 — AF3가 예측한 단백질-DNA, 항원-항체 복합체 예시. (c) 리간드·핵산·공유 변형·단백질 각 카테고리에서 AF-M 2.3, RFAA, AutoDock Vina 등 베이스라인과의 비교. (d) 전체 파이프라인: 입력이 Pairformer로 들어가고, 그 pair representation을 조건으로 Diffusion Module이 원자 좌표를 생성한다.
입력이 원자 단위로 통일됐다
AF2는 아미노산 잔기를 단위로 깔고 있었기 때문에 DNA·RNA·리간드를 넣을 자리가 원칙적으로 없었다. AF3는 잔기든 염기든 이온이든 소분자 원자든 전부 같은 토큰 공간으로 매핑한다. 타입 정보는 채널로 따로 주고, 연산은 공통이다.
작아 보이지만 이게 핵심이다. AF-Multimer나 RoseTTAFold All-Atom이 “단백질 + 확장 모듈” 조립식으로 풀던 걸, AF3는 표현 층에서 지워버렸다. 모델이 “단백질이 DNA를 만나는 자리”와 “단백질이 리간드를 만나는 자리”를 구분 없이 같은 경로로 다룬다.
Evoformer가 Pairformer로 얇아졌다
AF2는 MSA 표현과 pair 표현을 48블록에 걸쳐 둘 다 적극적으로 섞었다. AF3는 MSA의 지분을 줄이고 pair 중심으로 흐른다. 이건 타입을 통일한 순간부터 사실상 자연스러운 수순이다. MSA는 단백질 진화 정보인데, 이온이나 리간드엔 MSA라는 개념 자체가 없거나 약하기 때문에 그걸 중심 표현으로 둘 수 없다. MSA는 여전히 입력으로 들어오지만 초반에 pair로 응축시키고 뒷단은 pair 위주로 돌아간다.
Triangle multiplication/attention은 그대로 살아있다. 기하 제약(삼각 부등식)은 원자 단위에서도 맞는 얘기고, AF2 Evoformer에서 가장 영리했던 장치 중 하나가 여기서도 유효하다. “도메인 지식을 아키텍처에 심는다”는 AF2의 철학이 완전히 지워지진 않은 셈.
Structure Module이 Diffusion Module로 갈아 끼워졌다
AF2의 Structure Module은 회귀였다. pair representation에서 local frame들을 읽고, Invariant Point Attention으로 좌표를 한 번에 찍고, FAPE loss로 감독하고, 8번 iterate. 상당히 아름다운 설계였다.
AF3는 이 자리를 diffusion으로 바꾼다. 노이즈에서 출발해 원자 좌표를 점진적으로 정제하고, Pairformer의 pair representation을 조건으로 받는다.
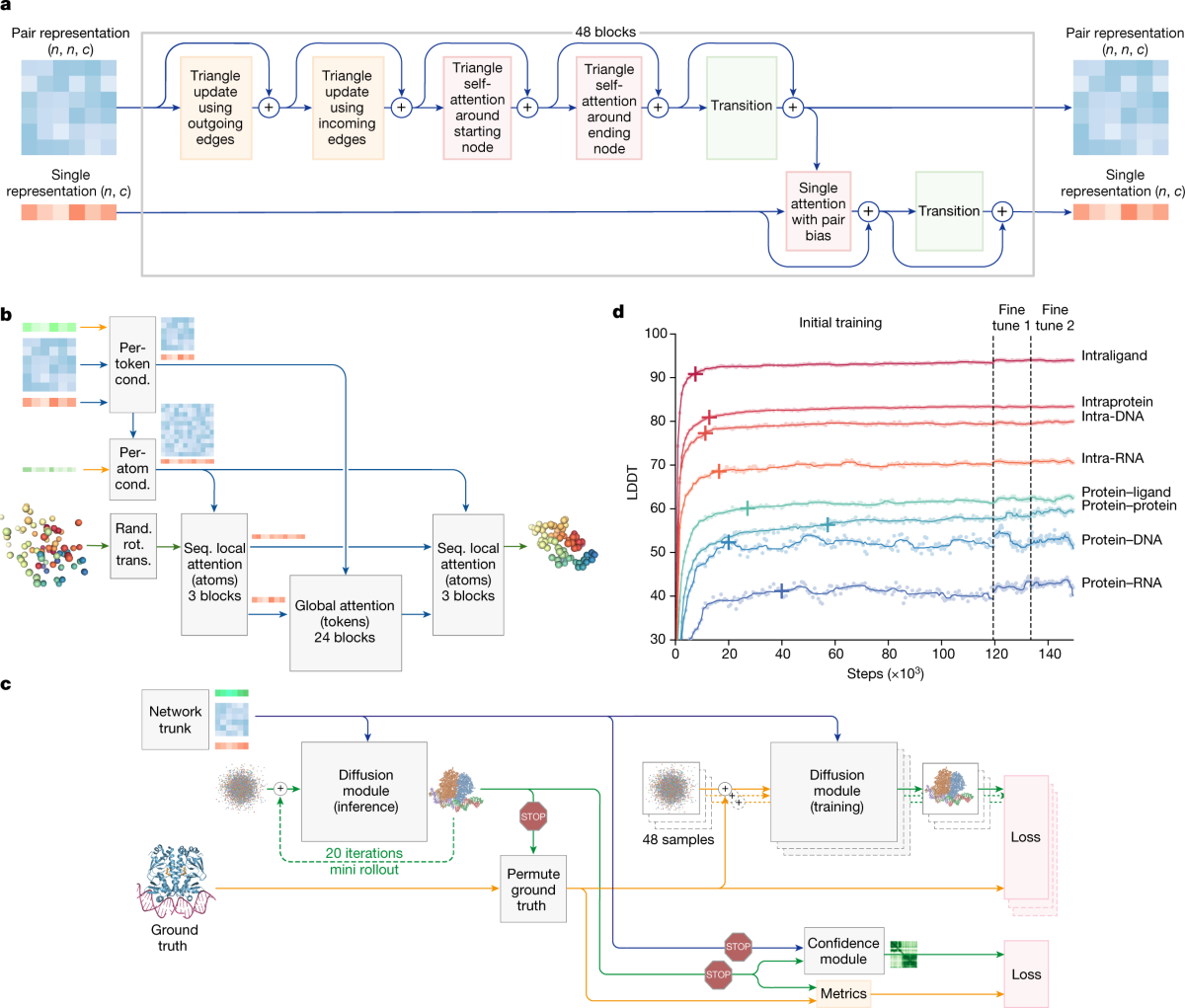 Fig. 2 (Abramson et al., 2024). (a) Pairformer 48블록 — triangle 연산이 pair representation을 업데이트하고 single representation이 함께 흐른다. (b) Diffusion module의 per-atom / per-token 2단 구조. 원자 단위 local attention으로 좁게 본 뒤, token 단위 global attention 24블록으로 복합체 전체를 묶는다. (d) 학습 곡선 — 단백질 내부 LDDT가 먼저 수렴하고, protein-ligand / protein-RNA 인터페이스가 뒤따라 올라온다.
Fig. 2 (Abramson et al., 2024). (a) Pairformer 48블록 — triangle 연산이 pair representation을 업데이트하고 single representation이 함께 흐른다. (b) Diffusion module의 per-atom / per-token 2단 구조. 원자 단위 local attention으로 좁게 본 뒤, token 단위 global attention 24블록으로 복합체 전체를 묶는다. (d) 학습 곡선 — 단백질 내부 LDDT가 먼저 수렴하고, protein-ligand / protein-RNA 인터페이스가 뒤따라 올라온다.
왜 회귀를 그만뒀을까. IPA가 전제하는 “잔기 frame(backbone 개념)”이 범용 복합체에 맞지 않기 때문이다. 리간드엔 backbone이 없고 이온은 단일 원자고 핵산은 backbone 모양이 다르다. AF2의 우아함은 단백질 전용이어서 우아했던 것이고, 그 우아함이 일반화의 족쇄가 된다. Diffusion으로 가면 원자를 그냥 점들로 놓고 점의 분포를 배우면 되니까 타입 불문으로 풀린다.
대가는 따로 적는다.
Recycling은 그대로다
Pairformer의 출력을 입력에 되넣는 recycling은 AF2와 같은 개념으로 유지. Diffusion 내부의 샘플링 스텝과는 다른 층위의 반복이라는 점만 짚어둘 만하다.
Diffusion이 가져온 새 실패 모드
Diffusion 계열 모델은 표현력 대신 “그럴듯하지만 틀린” 구조를 만드는 성향이 있고, AF3도 이걸 피해 가지 못했다. 저자들이 이 부분을 솔직하게 드러낸 게 오히려 논문의 신뢰도를 높인다.
가장 인상적인 실패 모드는 disordered region을 자기 확신에 차서 “접힌 모양”으로 그려내는 케이스다. Ground truth는 풀려있는 리본인데 AF3는 멀끔한 alpha helix로 예측한다. AF2는 같은 영역을 낮은 pLDDT로 “여긴 잘 모르겠다”고 표시하고 지나갔는데, AF3는 자신 있게 틀릴 때가 있다.
Stereochemistry(결합 길이·각도)와 chirality(D/L 이성질체) 오류도 간헐적으로 튀어나온다. AF2에서는 local frame이라는 구조적 제약이 이걸 거의 강제로 막았는데, diffusion은 통계적으로만 배울 수 있다.
이에 대한 완화책이 몇 개 붙어있다. chirality를 틀리면 페널티를 주는 보조 손실, bond length/angle에 페널티를 주는 물리 기반 손실, AF2의 예측을 추가 학습 데이터로 끌어오는 cross-distillation, 그리고 여러 샘플을 뽑아 confidence로 랭킹하는 inference 워크플로. 그럼에도 disordered region과 훈련 밖 리간드에서는 한계가 남는다.
AF2와 AF3의 가장 큰 철학적 차이가 여기다. AF2는 “가능한 구조를 좁혀서 하나를 찍는다”였고, AF3는 “여러 가능한 구조를 뽑아 고른다”에 가깝다. 후자는 유연하지만 위험하다.
결과 — 전용 도구를 한 모델이 제친다
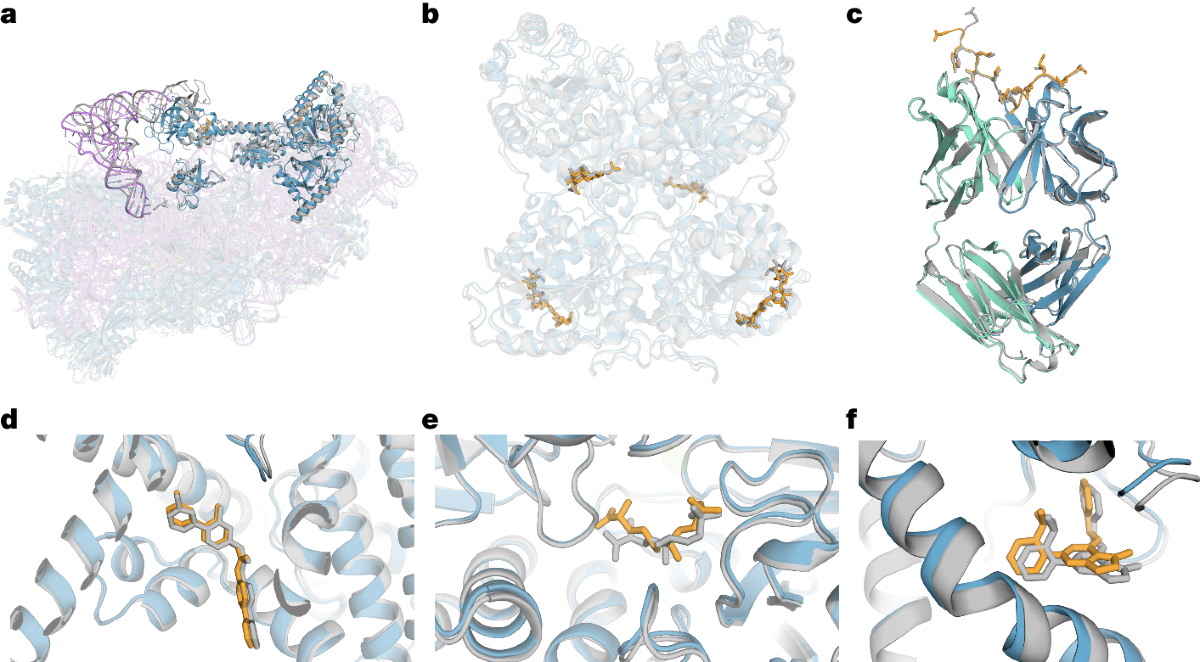 Fig. 3 (Abramson et al., 2024). AF3 예측(파란 리본)과 실험 구조(회색)를 겹친 예시들. 리간드 포즈(오렌지), 당 사슬, 인산화 잔기 등 AF2에선 모델링 자체가 불가능했던 요소들이 바인딩 포켓에 그대로 앉는다.
Fig. 3 (Abramson et al., 2024). AF3 예측(파란 리본)과 실험 구조(회색)를 겹친 예시들. 리간드 포즈(오렌지), 당 사슬, 인산화 잔기 등 AF2에선 모델링 자체가 불가능했던 요소들이 바인딩 포켓에 그대로 앉는다.
임팩트가 컸던 벤치마크는 PoseBusters(단백질-리간드 도킹)였다. AutoDock Vina, Gold 같은 수십 년 된 도킹 도구들을 범용 모델이 정확도에서 제쳤다는 게 약 개발 쪽에서 꽤 큰 파장이었다. 핵산 쪽에서도 RoseTTAFold All-Atom을 유의미하게 앞섰고, antibody-antigen은 AF-Multimer 2.3 대비 뚜렷하게 개선됐다. 공유 변형(인산화, 당화 등)은 지원 범위가 제한적이지만 지원되는 범위 안에선 잘 맞춘다.
“전문 도구 여러 개를 한 모델로 대체한다”는 주장은 대체로 성립한다. 완전한 대체는 아니지만.
신뢰도 쪽에서는 AF2에서 쓰던 pLDDT/PAE에 pTM(구조 전체 품질)과 ipTM(인터페이스 품질)이 붙었다. 복합체 시대에 없으면 안 되는 지표다. “단백질은 잘 접혔는데 리간드 포즈는 불확실하다” 같은 구분이 한 모델 안에서 가능해진 게 실무적으론 생각보다 크다.
한계 — 저자가 쓴 것 + 써보면서 보이는 것
저자들이 직접 인정한 쪽부터 쓰면, disordered region 쪽의 hallucination, stereochemistry/chirality 간헐적 오류, 여전히 정적 구조 하나라는 점(알로스테릭 전환 못 잡음, AF2와 공유하는 한계), 오픈소스 버전에서 지원하는 리간드·변형이 좁다는 점, 그리고 glycan 커버리지 부족이 있다. 인간 프로테옴의 상당 부분이 당 사슬 없이는 기능적으로 불완전한데 AF3 지원은 부분적이라서, “인간 단백질 구조를 다 보여준다”는 말에는 아직 괄호가 붙는다.
후속 연구에서 드러난 것들도 무시하기 어렵다. 훈련 세트와 유사도가 낮은 리간드에서 정확도가 급격히 떨어진다는 보고가 여러 번 나왔고, binding pocket의 물리화학적 특성을 의도적으로 흔들어 만든 adversarial decoy에 AF3가 둔감하다는 연구도 있다. 정말로 화학을 이해한 게 아니라 훈련 데이터를 기억했을 가능성이 남는다는 뜻. 그리고 diffusion인 이상 seed에 따라 출력이 바뀐다. 여러 샘플을 뽑고 confidence로 거르는 과정이 사실상 필수적인 워크플로가 된다.
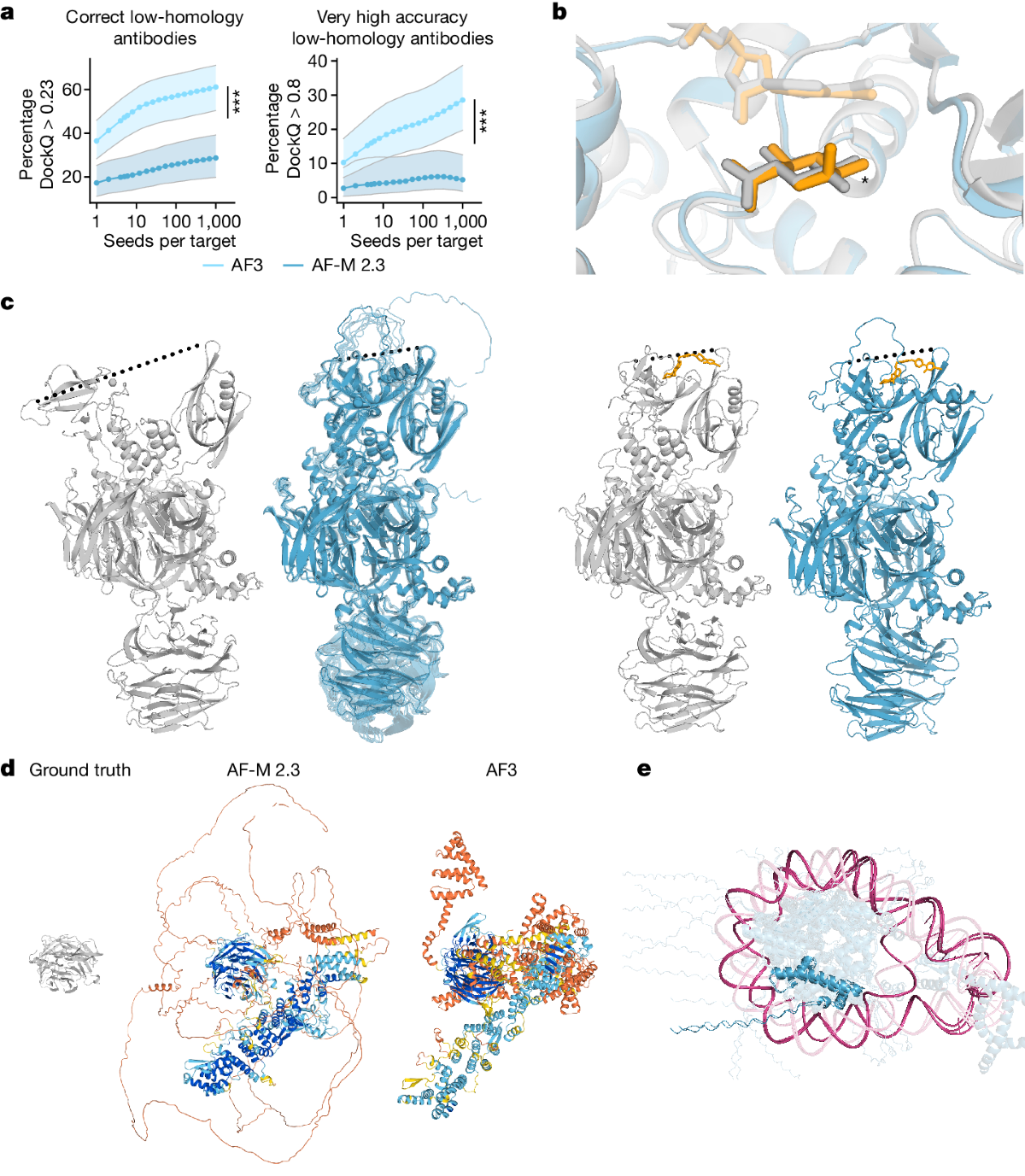 Fig. 5 (Abramson et al., 2024). (a) Antibody-antigen 인터페이스는 seed(샘플) 수를 늘릴수록 성공률이 단조 증가한다. AF-Multimer 2.3 대비 개선이 뚜렷하지만, 바꿔 말하면 “맞는 게 섞여 나온다”는 성격도 갖는다. (c–d) AF3가 disordered region을 자신 있게 접힌 모양으로 그려낸 대표적 hallucination 예시.
Fig. 5 (Abramson et al., 2024). (a) Antibody-antigen 인터페이스는 seed(샘플) 수를 늘릴수록 성공률이 단조 증가한다. AF-Multimer 2.3 대비 개선이 뚜렷하지만, 바꿔 말하면 “맞는 게 섞여 나온다”는 성격도 갖는다. (c–d) AF3가 disordered region을 자신 있게 접힌 모양으로 그려낸 대표적 hallucination 예시.
Fig. 5(a)가 상징적이다. seed 수가 늘수록 성공률이 올라간다. 좋게 보면 스케일링 성질이 있다는 뜻이고, 뒤집어 보면 한 번에 답을 얻는 구조가 아니라 여러 번 돌려서 고르는 구조라는 뜻이다.
라이선스 얘기 — 생각보다 크다
AF2가 Apache 2.0이었던 것에 비해 AF3는 CC-BY-NC-SA 4.0이다. 비상업, share-alike. 가중치는 2024-11에 초대 기반으로 먼저 풀렸다가 2025-02에 일반 공개로 전환됐지만 라이선스는 그대로다. AlphaFold Server는 비상업 연구 한정 무료. 상업 파이프라인은 Isomorphic Labs가 쥐고 있고, 외부 접근은 닫혀 있다.
학계 연구에는 문제가 없지만, 신약 개발 스타트업이 AF3를 그대로 제품 파이프라인에 꽂는 건 불가능하다. AF2 시대처럼 생태계가 폭발적으로 확산되지 않는 이유가 여기에 있다.
마지막으로
한 줄로 말하고 싶지는 않은데, 굳이 쓰자면 AF2가 단백질의 물리를 네트워크 구조에 새긴 모델이었다면 AF3는 그 구조적 bias를 풀고 표현력으로 간 모델이다. IPA·FAPE·frame이 단백질에 강했던 만큼 그 바깥에선 안 펴졌고, AF3는 그 제약을 들어내는 대가로 복합체 전반에서 통합된 예측을 얻었다. 잃은 건 물리적 일관성의 강제력과 해석의 안정성이고, 그게 곧 hallucination이다.
지금 쓰기 좋은 자리는 어쨌든 명확하다. 약 개발의 초기 트리아지, 단백질-핵산 복합체 스케치, 공유 변형이 있는 단백질, antibody-antigen 후보 필터링. 대신 정밀 약물 설계에서 훈련 밖 리간드를 다룰 때, disordered region 해석이 필요할 때, 그리고 상업 파이프라인에 꽂을 때는 조심해야 한다. 전용 도구를 대체하려 쓰기보단 앞단에 붙여 쓸 때 제일 잘 맞는다. 그 정도 거리감이 지금은 맞는 것 같다.